
Abstract
Gene expression is the molecular language of biology, and Ribonucleic Acid Sequencing (RNA-Seq) is how we read it. Since its introduction, RNA-Seq has displaced micro arrays, redefined transcriptomics, and become the standard assay for understanding what genes are active, in which cells, under what conditions, and by how much. Yet for many researchers, the journey from raw FASTQ files to a figure ready for Nature or Cell remains opaque, fragmented, and riddled with tool choice anxiety.
This tutorial closes that gap. Written for bench scientists, graduate students, clinical researchers, and bioinformatics beginners, it walks through every stage of a bulk RNA-Seq workflow quality control, read alignment, gene quantification, differential expression analysis, and biological pathway interpretation. Each step reflects the validated, reproducible BioCompute NGS pipeline used by Genix.ai, where PhD level analysts run these exact tools for research teams across pharma, biotech, academia, and clinical genomics, delivering publication ready results in 3 to 5 business days.
Whether you are running the pipeline yourself or evaluating what a professional bioinformatics pipeline development service should deliver, this guide gives you the full picture.
What Is RNA Seq And What Does the Name Actually Mean?
RNA-Seq stands for Ribonucleic Acid Sequencing, sometimes written simply as RNA Sequencing, where "Seq" is a standard shorthand for sequencing in the genomics field.
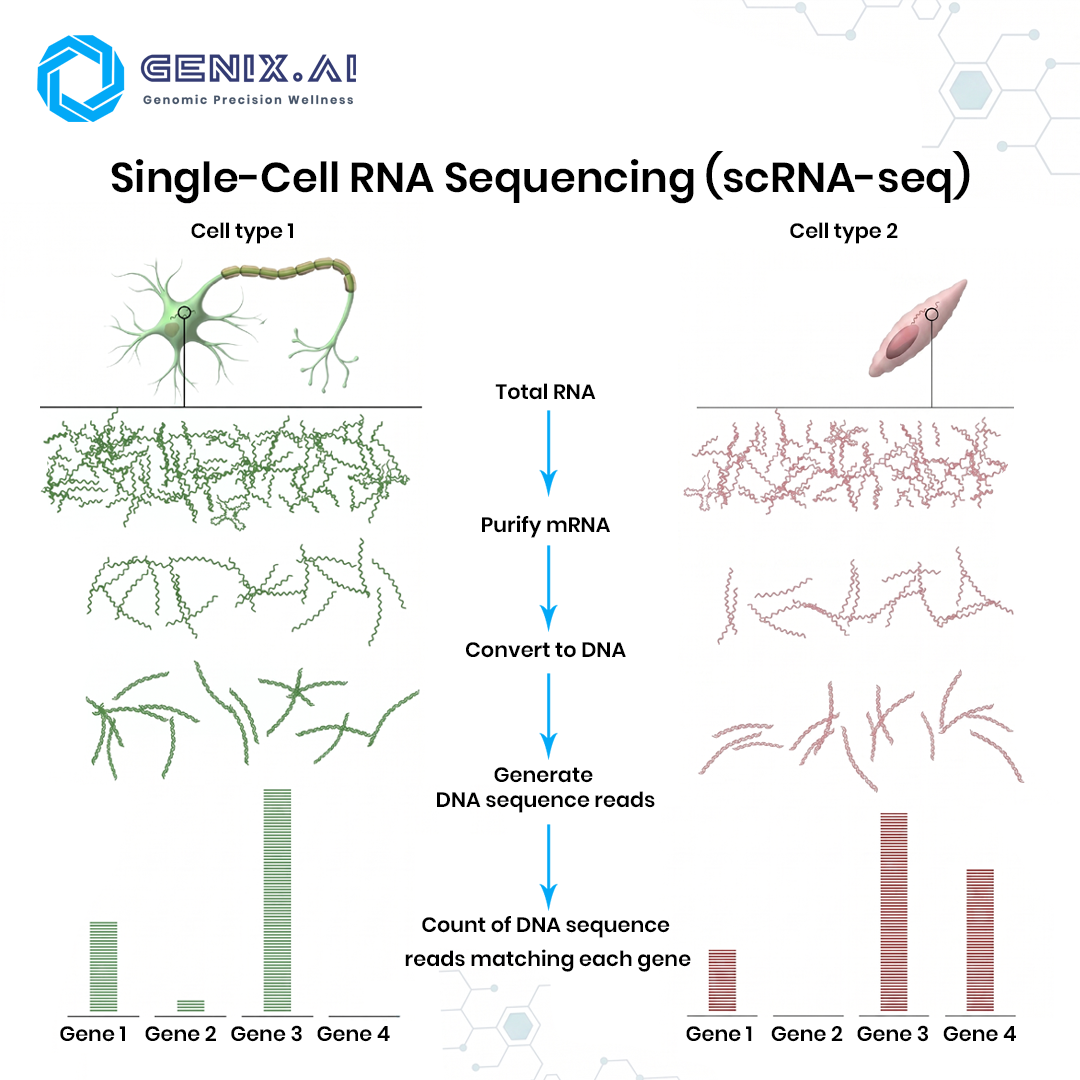
RNA-Seq is a method for sequencing an entire set of RNA molecules. It involves isolating RNA molecules from a tissue or cell sample, transforming those RNAs into DNA, and then sequencing the resulting DNA molecules. RNA-Seq helps researchers determine which segments of DNA have been transcribed into RNA and how much a gene is expressed.
While two different cells within the human body have a similar or identical set of DNA molecules in the genome, the set of RNA molecules, called the transcriptome, varies significantly between cells. A skin cell and a brain cell each contain different RNA. Unlike DNA, the RNA within a given cell is dynamic, rapidly changing over time based on many factors from time of day to diet, or in more extreme cases, whether that cell is healthy or diseased.
This dynamic nature is exactly why RNA-Seq has become indispensable in disease research, drug development, and clinical genomics.
What Is FASTQ : The Full Form and Why It Matters?
FASTQ stands for FASTA + Quality : a combination of two concepts:
- FASTA is derived from FAST All, a DNA and protein sequence alignment software that introduced the plain text sequence file format.
- Q stands for Quality, specifically, per base Phred quality scores that indicate the confidence of each nucleotide call made by the sequencer.
A FASTQ file is a text based file format that stores both a raw nucleotide sequence and its corresponding per base quality scores, output directly by every NGS sequencer on the market. Every RNA-Seq experiment begins here. A single paired end sample generates two FASTQ files, one for each read direction, and the quality of everything downstream depends entirely on what is inside them. A contaminated, adapter heavy, or low quality FASTQ file produces unreliable counts, misleading differential expression results, and figures that will not survive peer review.
Understanding what a FASTQ file contains and how to evaluate it is the single most important foundational skill in RNA-Seq bioinformatics pipeline development.
The Types of RNA: What RNA Seq Actually Captures
Not all RNA is the same. RNA-Seq can be designed to capture different RNA species depending on the biological question being asked. Understanding the major types of RNA clarifies why library preparation choices matter and which pipeline steps apply to your experiment.
Messenger RNA (mRNA) is the most commonly sequenced RNA type. It carries genetic instructions from DNA to the ribosome for protein synthesis, and is the primary target of standard bulk RNA-Seq experiments. Most gene expression studies measuring which genes are up regulated or down regulated are built around mRNA.
Ribosomal RNA (rRNA) constitutes over 80% of total cellular RNA but carries no gene expression information relevant to most experiments. It is actively depleted during library preparation using ribo depletion kits or poly-A selection, which enriches for mRNA. Failing to remove rRNA wastes sequencing depth and distorts results.
Transfer RNA (tRNA) plays a structural role in translation, but is rarely the target of standard RNA Seq. Dedicated small RNA protocols are required to capture it effectively.
Long Non Coding RNA (lncRNA) is a rapidly expanding class of RNA molecules longer than 200 nucleotides that do not encode proteins but regulate gene expression, chromatin remodeling, and cellular identity. lncRNA focused RNA Seq requires ribo depletion rather than poly-A selection, as many lncRNAs lack a poly-A tail.
Small Non Coding RNAs including microRNA (miRNA), small interfering RNA (siRNA), and small nucleolar RNA (snoRNA) regulate post transcriptional gene expression and require specialized small RNA sequencing protocols with distinct pipeline parameters.
Circular RNA (circRNA) is a closed loop RNA form increasingly implicated in disease biology and biomarker research, detectable through specific alignment strategies that account for back splicing junctions.
Knowing which RNA type your experiment targets determines your library preparation kit, your ribo depletion or enrichment strategy, your alignment reference, and which steps of the bioinformatics pipeline development workflow apply. Genix.ai's BioCompute team confirms RNA type and library design at the project intake stage,before a single command is run.
The RNA -Seq Bioinformatics Pipeline Development Workflow
A standard bulk RNA -Seq experiment follows a linear, validated sequence of computational steps. Raw reads enter the pipeline as FASTQ files. Each stage transforms that data by cleaning, mapping, counting, testing statistically, and finally translating numbers into biological insight.
The tool chain used in Genix.ai's BioCompute bioinformatics pipeline development service mirrors what top tier institutions publish with:
FastQC + MultiQC → Trimmomatic or fastp → STAR → Salmon or feature Counts → DESeq2 or edgeR → cluster Profiler → publication figures. Every tool is production tested, version controlled, and part of a reproducible workflow, not a loose collection of scripts.
Step by Step: From FASTQ to Publication Ready Results
Step 1 Quality Control: Interrogate Your Raw Data First

The most expensive mistake in RNA-Seq is skipping quality control. Raw FASTQ files carry noise: adapter sequences, low-quality base calls at read ends, GC content bias, and PCR duplicates. Run FastQC on every file before touching a single downstream tool.
bash
fastqc sample_R1.fastq.gz sample_R2.fastq.gz -o ./fastqc_output/Consolidate all sample QC reports using MultiQC into one interactive HTML dashboard, essential when working with 20 or more samples.
bash
multiqc ./fastqc_output/ -o ./multiqc_report/Inspect four things: per base quality scores (Phred ≥ 30 across most positions), adapter contamination, duplication rate, and GC distribution. Any abnormality here is a signal to investigate the library preparation, not paper over it downstream. Genix.ai's BioCompute team delivers a full QC report at this stage as the first formal project deliverable.
Step 2 Read Trimming: Remove What Does Not Belong
Quality control tells you what is wrong. Trimming fixes it. Use Trimmomatic for fine-grained control or fastp for speed and an integrated HTML report.
fastp -i sample_R1.fastq.gz -I sample_R2.fastq.gz \
-o sample_R1_trimmed.fastq.gz -O sample_R2_trimmed.fastq.gz \
--detect_adapter_for_pe \
--cut_tail --cut_mean_quality 20 \
--html fastp_report.html After trimming, run FastQC again and confirm improvement. Clean reads are the non-negotiable foundation of any reproducible bioinformatics pipeline development effort.
Step 3 Alignment: Map Reads to the Reference Genome
Align trimmed reads to a reference genome using STAR, the splice-aware aligner of choice for eukaryotic RNA-Seq. For human data, use GRCh38 with the corresponding Ensembl GTF annotation.
STAR --runMode genomeGenerate \
--genomeDir /ref/hg38_STAR_index \
--genomeFastaFiles GRCh38.primary_assembly.genome.fa \
--sjdbGTFfile Homo_sapiens.GRCh38.110.gtf \
--runThreadN 16STAR --runMode alignReads \
--genomeDir /ref/hg38_STAR_index \
--readFilesIn sample_R1_trimmed.fastq.gz sample_R2_trimmed.fastq.gz \
--readFilesCommand zcat \
--outSAMtype BAM SortedByCoordinate \
--outSAMattributes NH HI AS NM \
--runThreadN 16 \
--outFileNamePrefix ./aligned/sample_Target alignment rates for human samples are 85–95%. Rates below 70% indicate contamination or severe library quality issues, all of which Genix.ai's pipeline flags before proceeding.
Step 4 Quantification: Count Reads Per Gene
Use Salmon for its speed, accuracy, and built-in bias correction for GC content and fragment length.
salmon quant -i /ref/salmon_index \
-l A \
-1 sample_R1_trimmed.fastq.gz \
-2 sample_R2_trimmed.fastq.gz \
--validateMappings \
--gcBias \
--threads 16 \
-o ./quant/sample/The output is a per-gene count matrix, the input to all statistical analysis. Genix.ai delivers this normalized count matrix as a standalone deliverable, giving you full downstream flexibility.
Step 5 Differential Expression: Identify What Changes and Why
Load the count matrix into DESeq2 in R, define your experimental design, and run the statistical model
r
library(DESeq2)
counts <- read.csv("count_matrix.csv", row.names = 1)
metadata <- read.csv("sample_metadata.csv", row.names = 1)
dds <- DESeqDataSetFromMatrix(countData = counts,
colData = metadata,
design = ~ condition)
dds <- dds[rowSums(counts(dds)) >= 10, ]
dds <- DESeq(dds)
res <- results(dds, contrast = c("condition", "treatment", "control"))
sig <- subset(res, padj < 0.05 & abs(log2FoldChange) > 1)Visualise with a PCA plot, volcano plot, and heatmap. These three figures, polished to journal standards, are standard in every Genix.ai BioCompute RNA-Seq delivery.
Step 6 Pathway Enrichment: Translate Statistics into Biology
Use cluster Profiler for GO and KEGG enrichment, then run GSEA on a ranked gene list.
r
library(clusterProfiler)
library(org.Hs.eg.db)
ego <- enrichGO(gene = rownames(sig),
OrgDb = org.Hs.eg.db,
keyType = "SYMBOL",
ont = "BP",
pAdjustMethod = "BH",
qvalueCutoff = 0.05)
dotplot(ego, showCategory = 20)
ekegg <- enrichKEGG(gene = entrez_ids,
organism = "hsa",
pvalueCutoff = 0.05)
barplot(ekegg, showCategory = 15)Dotplots, barplots, and enrichment maps with KEGG, GO, and Reactome annotations are standard output in Genix.ai's BioCompute reports.
Conclusion: Why Genix.ai BioCompute Is the Smartest Next Step
Running this bioinformatics pipeline development workflow end to end requires version managed software environments, substantial compute (STAR indexing alone needs 32 GB RAM), and the scientific judgment to distinguish biologically meaningful results from technical artifacts. That is a significant investment for a lab whose primary expertise is biology, not infrastructure.
Genix.ai's BioCompute NGS Analysis service handles the entire pipeline described in this tutorial FastQC through cluster Profiler with PhD level review on every deliverable. Upload your FASTQ files, specify your conditions, and receive a normalized count matrix, DE gene list, volcano plots, pathway enrichment figures, and a copy paste ready methods section written to Nature/Cell formatting conventions all within 3 to 5 business days, starting at $150 per sample.
For pharma and biotech teams scaling to 50+ samples, volume pricing drops as low as $150 per sample with priority queue access. For academic researchers, student and institutional pricing is available. A full NDA covers every project, IP ownership, and data deletion on request. The Genix.ai BioCompute team built this service specifically so that the bioinformatics never blocks the science.
5 FAQs
1. What does RNA-Seq stand for and what does it measure?
RNA Seq stands for Ribonucleic Acid Sequencing, it measures which genes are expressed in a biological sample and at what level.
2. What is a FASTQ file, and why does every RNA-Seq pipeline start with it?
FASTQ (FASTA + Quality) is the raw output file from every NGS sequencer, storing nucleotide sequences alongside per base quality scores.
3. What are the main types of RNA captured by RNA-Seq?
The major types include mRNA, lncRNA, miRNA, rRNA, tRNA, and circRNA, each requiring different library preparation and pipeline strategies.
4. Why is bioinformatics pipeline development critical for publication ready RNA-Seq results?
A validated, version controlled pipeline ensures reproducibility, correct tool parameters, and results that survive both peer review and replication.
5. Can Genix.ai handle the entire RNA-Seq pipeline from FASTQ to publication figures?
Yes, Genix.ai's BioCompute NGS service delivers QC, alignment, DE analysis, pathway enrichment, figures, and methods text in 3 to 5 days from $150 per sample.





